
Research activities
Molecular Property Diagnostic Suite Software (MPDS)
The Molecular Property Diagnostic Suite (MPDS) is conceptualized to assess and estimate the multifarious aspects of any given molecule, in order to diagnose their potential application as drug, material, catalysts, pesticide, perfume etc. The MPDS platform for a given disease, besides providing molecular modelling tools, chemo- and bioinformatics data, also combines genetic, epigenetic, pharmacogenomic and metabolomics data. MPDS consists of disease-independent and disease-dependent modules which are structured into data library (target library, compound library, fragment library, literature, gene library and pathway information), data processing (file format conversion, descriptor calculation, statistical tools and machine learning tools), data analysis (QSAR, docking, drug-likeness filter, and visualization tool, biological potential prediction) and advanced modules (disease-disease interaction, big data analytics in chemistry and healthcare, predictive computing and multi-scale modelling). Click on the image for more details.
Webportals
MPDSCOVID-19 Web Portal
The MPDS COVID-19 is a disease specific web portal for COVID-19. The portal has exhaustive information of COVID-19 that includes genes, proteins, pathways, variants etc. The portal also contains the results and outcomes of our study from drug repurposing, protein-protein interaction studies. The MPDS COVID-19 has new modules for pharmacophore model generation, binding site prediction and machine learning calculations. Click on the image for more details.

MPDSTB Web Portal
The MPDS-TB is a disease-specific open source web-portal which is a repository of all informations on tuberculosis. Chemoinformatics, CADD tools, and other computational techniques are provided on the single web-portal where each compound is identified with a unique MPDS ID number and an MPDS ID card is produced similar to Aadhar card in India. Click on the image for more details.
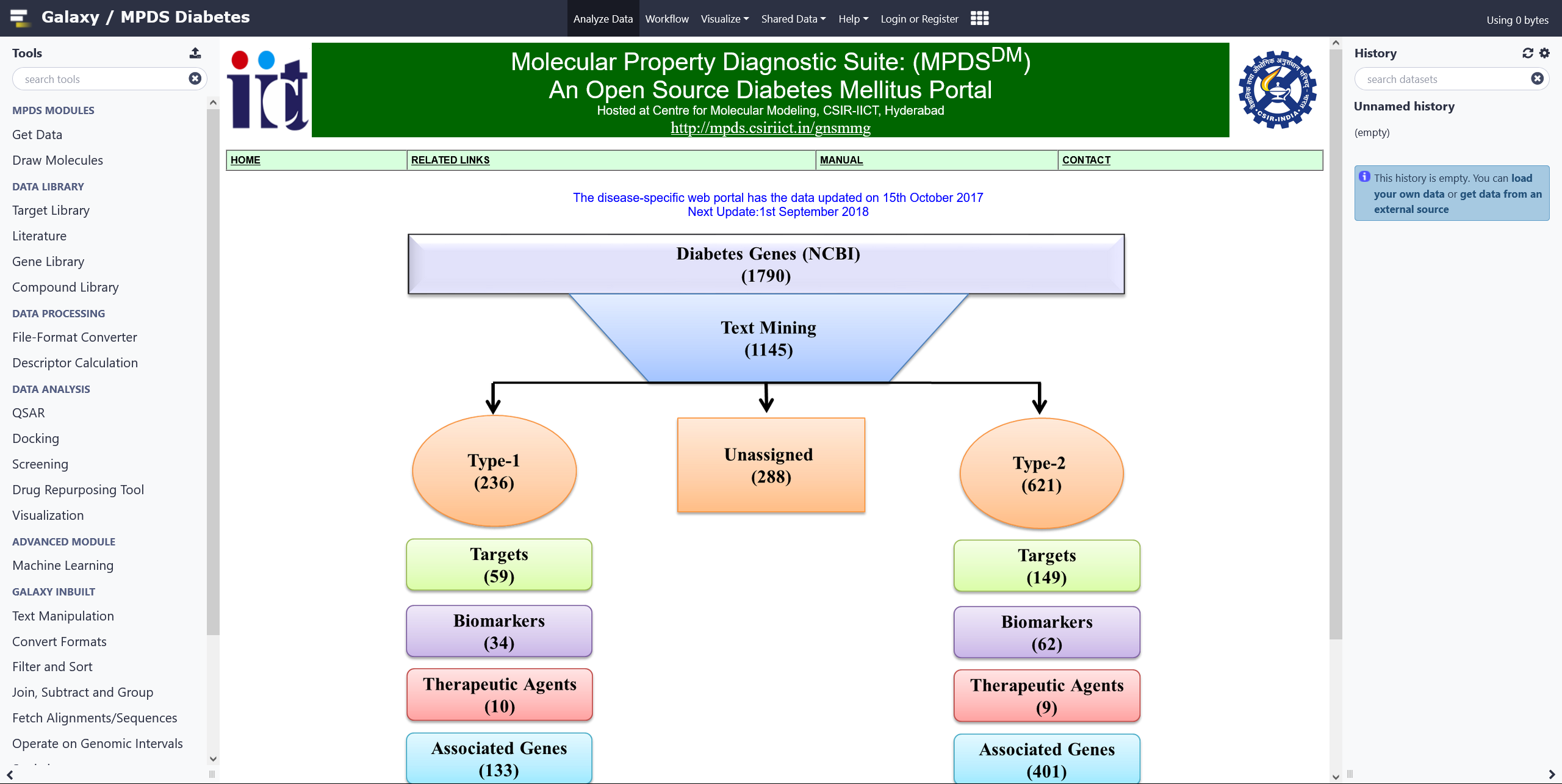
MPDSDM Web Portal
The MPDS-DM is a disease-specific open source web-portal which is a repository of all informations on Diabetes Mellitus. Click on the image for more details.
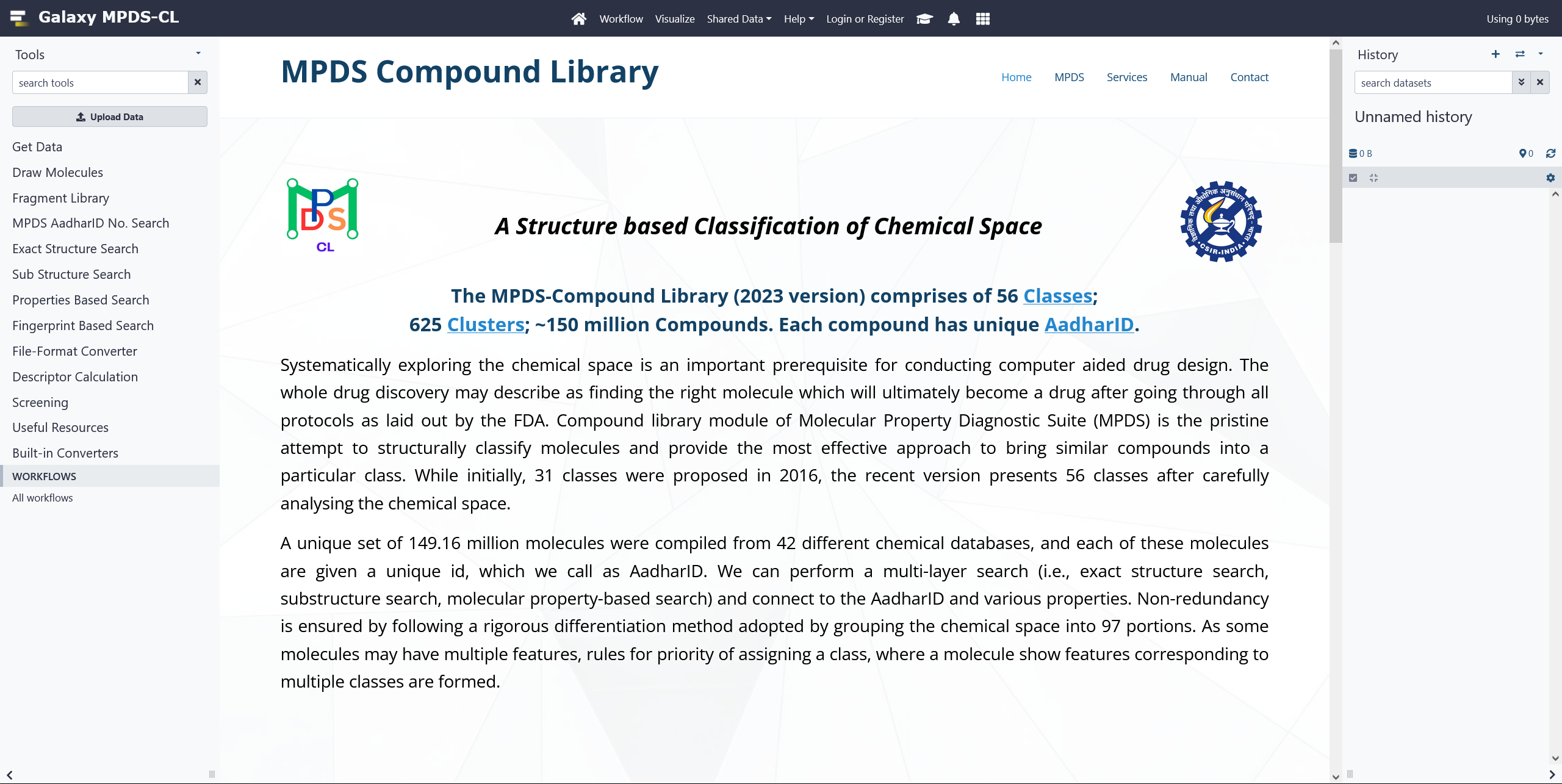
MPDSCL Web Portal
Systematically exploring the chemical space is an important prerequisite for conducting computer aided drug design. The whole drug discovery may describe as finding the right molecule which will ultimately become a drug after going through all protocols as laid out by the FDA. Compound library module of Molecular Property Diagnostic Suite (MPDS) is the pristine attempt to structurally classify molecules and provide the most effective approach to bring similar compounds into a particular class. While initially, 31 classes were proposed in 2016, the recent version presents 56 classes after carefully analysing the chemical space. A unique set of 149.43 million molecules were compiled from 42 different chemical databases, and each of these molecules are given a unique id, which we call as AadharID. We can perform a multi-layer search (i.e., exact structure search, substructure search, molecular property-based search) and connect to the AadharID and various properties. Non-redundancy is ensured by following a rigorous differentiation method adopted by grouping the chemical space into 97 portions. As some molecules may have multiple features, rules for priority of assigning a class, where a molecule show features corresponding to multiple classes are formed. Click on the image for more details.
Webportals (Under Development)
MPDSMD Web Portal
The MPDS-MD is a Disease-specific Web Portal for Metabolic Disorders which is another addition to the MPDS and currently it is under development.Click on the image for more details.


MPDSHIV Web Portal
The MPDS-HIV is a Disease-specific Web Portal for Human Immuno Deficiency Virus which is another addition to the MPDS and currently it is under development.
Online Databases
OSADHI
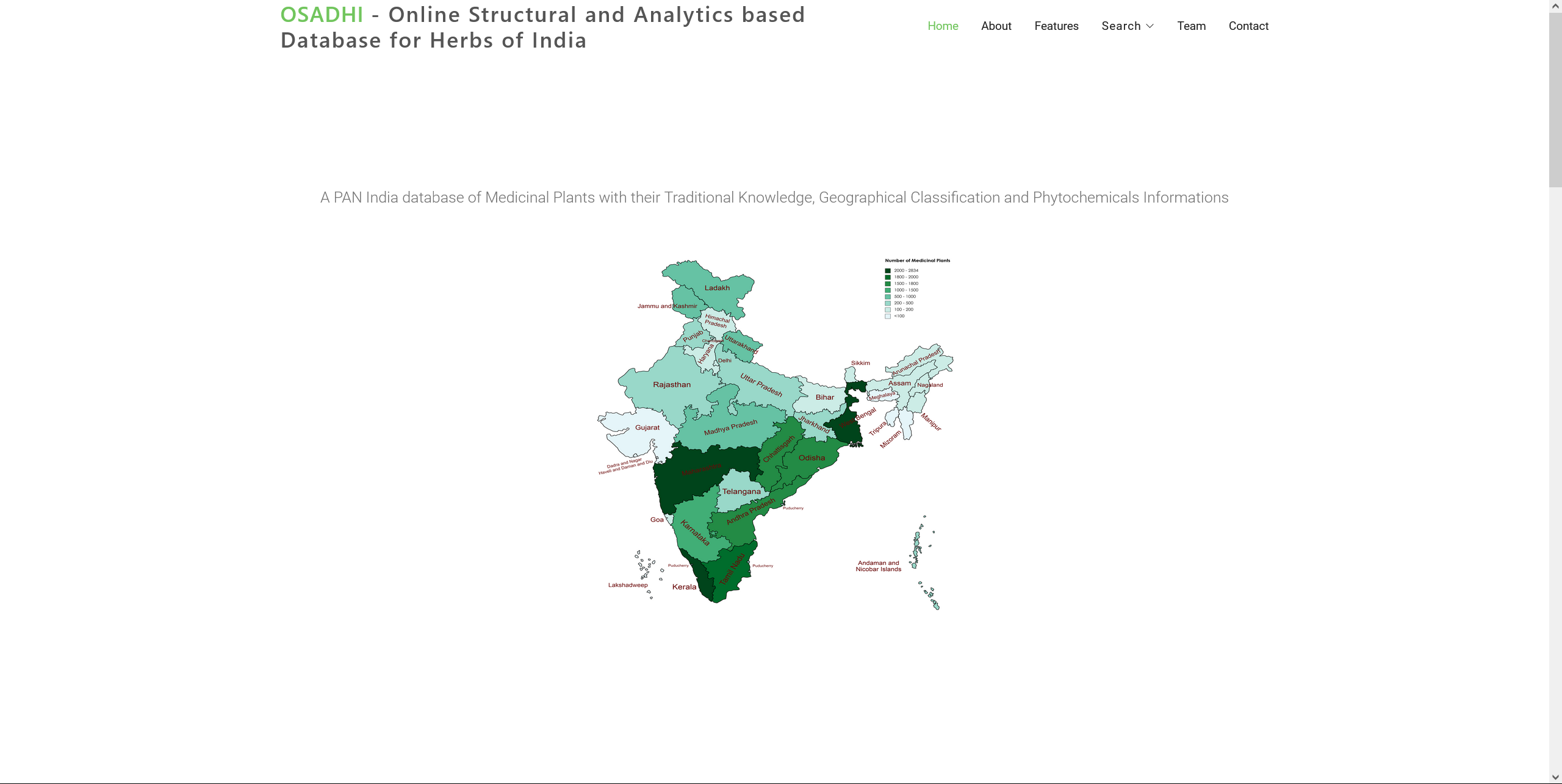
OSADHI - Online Structural and Analytics based Database for Herbs of India is one of the largest manually curated databases which have been constructed with the concept of "4Ds - Documentation, Digitization, Deposition and Data Science". The information in this database has been collected from books, published articles other existing open sources/repository. OSADHI consists of four major features -
- Traditional Knowledge - Information on vernacular names, plants parts used, therapeutic use and taxonomy.
- Geographical Classification - Distribution of medicinal plants available across the Indian states and Union Territories.
- Phytochemicals - Details of the phytochemicals available in the medicinal plants like SMILES, InChIKey, IUPAC, 2D & 3D Structures.
- Chemoinformatics analysis - Information on physiochemical properties, ADMET, classification etc.
The current version of OSADHI has identified 21,238 medicinal plants available all over India out of which 6959 are unique. For these unique plants, 27,440 phytochemicals and 2,477 therapeutic uses have been captured along with the geographical locations. Along with vernacular names, 343 plant families have been reported in this database.Click on the image for more details.

NEIMPDB
NEI-MPDB is an integrated database of extensive list of medicinal plants used in the North Eastern part of India. The database has been carefully constructed by the various techniques, such as literature mining, text mining and manual curation. The database provides the following resources to users:
- Medicinal plants list specific to each state of North East India.
- Medicinal plants and the parts, method of use, and ailments cured.
- The phytochemical constituents of the medicinal plants.
The obtained list of plants was carefully curated by removing the redundant files and validation of input data. The unique plant list was selected and used for the development of the database. The database comprises of more than 550 plant list in addition to 9000+ phytochemicals. The development of this platform is an effort to help record the dying traditional practices and revive the age old practices. This will further intensify the computational studies on natural product drug discovery with the focus on North East India.Click on the image for more details.
A2ID
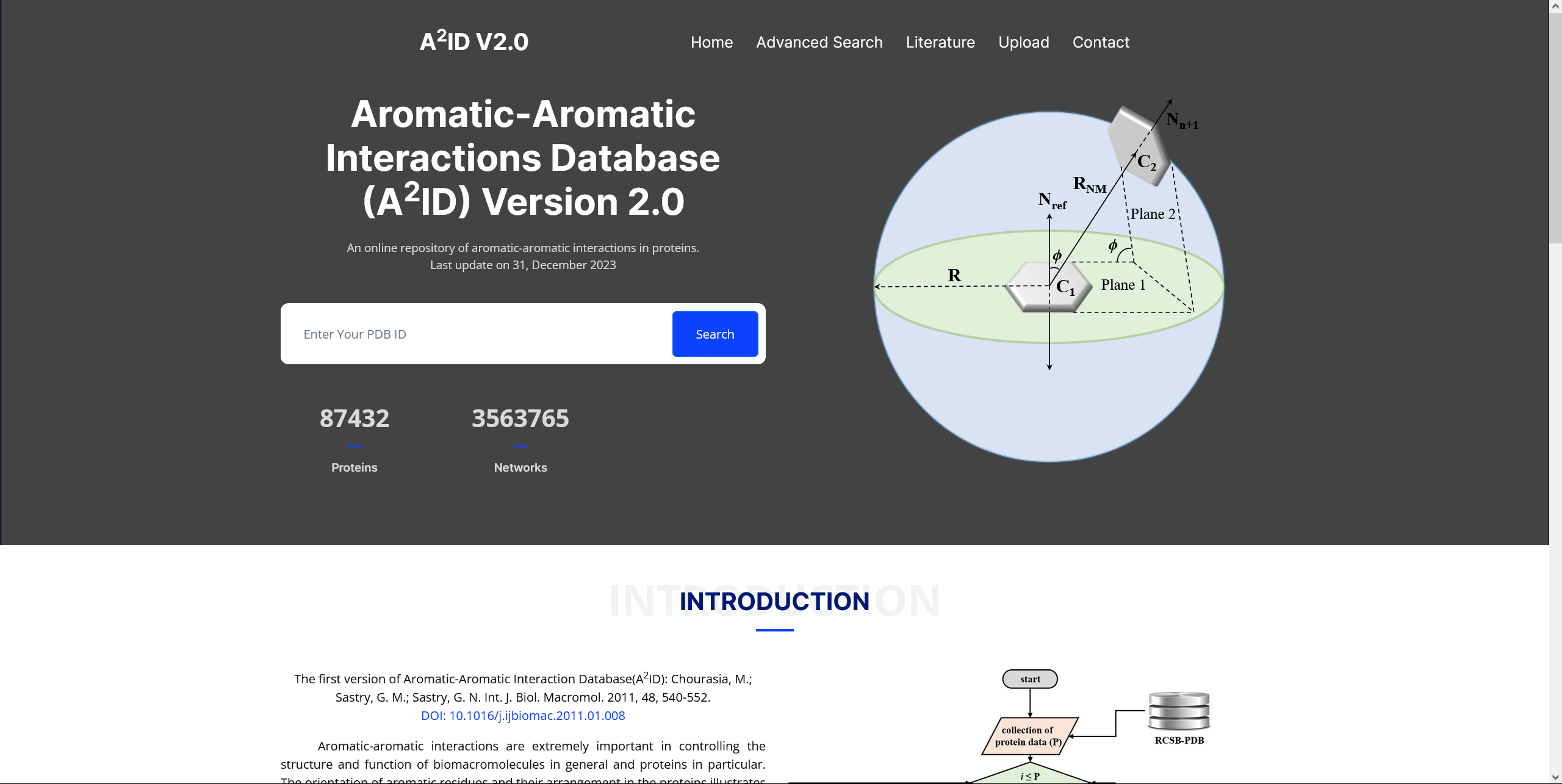
Aromatic-aromatic interactions are extremely important in controlling the structure and function of biomacromolecules in general and proteins in particular. The orientation of aromatic residues and their arrangement in the proteins illustrates the hidden cache responsible for structural stability. To the best of our knowledge we haven't come cross any systematic analysis on the occurrence of π-π motifs at a database level. Here we ventured on undertaking highly systematic approach to estimate the occurrence of π-π motifs in proteins. Geometrical analysis of adjacent aromatic rings of tryptophan (Trp), tyrosine (Tyr), phenylalanine (Phe), and histidine (His) of protein database was done. For this purpose five different classifiers SCOP (Structural Classification of Proteins), CATH (Class (C), Architecture (A), Topology (T) and Homologous superfamily (H)), EC (Enzyme Classification), SCOP2 (SCOP version2), and ECOD (Evolutionary Classification Of protein Domains) were considered. Click on the image for more details.
CAD V2.0
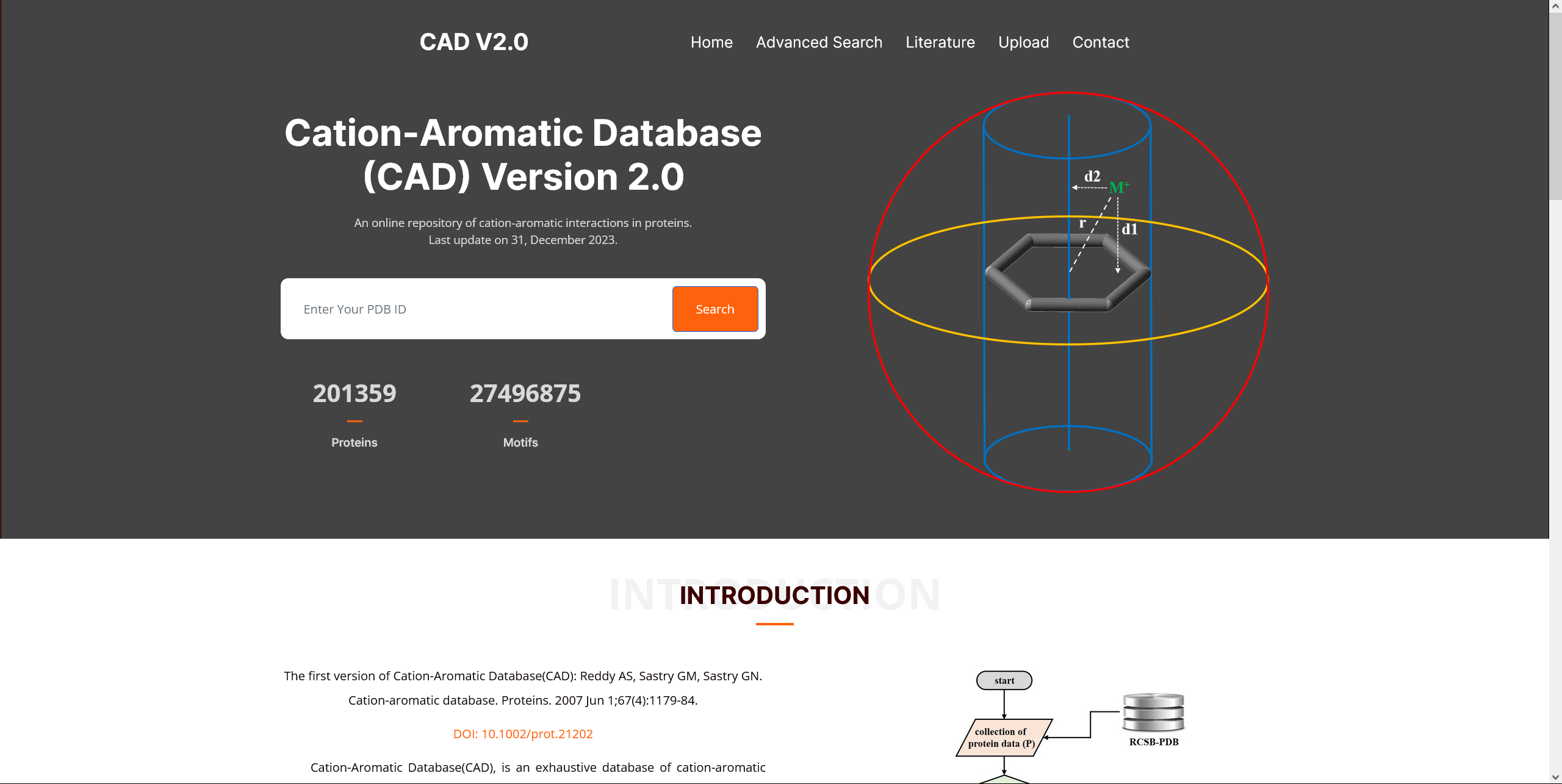
Cation-Aromatic Database(CAD), is an exhaustive database of cation-aromatic motifs that are present in the experimentally determined 3D structure of proteins, available in the protein data bank (PDB). The database contains statistical data about geometric arrangements, residue preferences, and interaction modes of all the underlying cationic and aromatic moieties, which include cation-σ and cation-π motifs. It aims to advance the knowledge of spatial distributions of cationic and aromatic residues and their interactions in protein structures, as well as how cation-aromatic motifs contribute to protein structure and function. Moreover, CAD has the potential to improve our ability to identify cation-π interactions quickly and facilitate the development of new methods and force fields. The updated version of CAD (CAD 2.0) includes information of ~27.26 million cation-aromatic motifs identified in the 3D structures of proteins. CAD identifies the position of the cation relative to the arene using three distance parameters r, d1, and d2, and classifies cation-aromatic motifs as either cation-π or cation-σ motifs.Click on the image for more details.
Machine Learning Tools
Antiviral Prediction using Machine Learning
The antiviral prediction models are based on CAS dataset published by American Chemical Society. This is a part of the disease independent module of Molecular Property Diagnostic Suite. An attempt has been made to develop Machine Learning (ML) based predictive models to identify potent antivirals compounds. A set of 2358 antiviral compounds were compiled from the CAS COVID-19 antiviral SAR dataset whose activity was reported based on IC50 value. A total 1157 2D molecular descriptors were computed and the most highly correlated descriptors were selected using different feature selection methods. Seven ML algorithms were benchmarked among which the best performance was achieved by the models developed using Random Forest and XGBoost algorithms in all the feature selection methods. The maximum predictive accuracy of both these models was 0.88 with internal validation. Whereas, with an external dataset, a maximum accuracy of 0.93. for XGBoost and 1.0 for Random Forest based model was achievable.Click on the image for more details.


Clinical Predictor using Machine Learning
The clincal trial predictor has been developed to assess the success and failure of a compound in clinical trials. Different machine learning algorithms have been used to make this tool with descriptors and fingerprints (1024 bit and 2048 bit) as features. The models have an accuracy of 70-72%. This is a part of the disease independent module of Molecular Property Diagnostic Suite. Our quest for the current study is to find out various reasons as to why an investigational candidate would fail in the clinical trials after multiple iterations of refinement and optimization. We have compiled a dataset that comprises of approved and withdrawn drugs as well as toxic compounds and essentially have used time-split based approach to generate the training and validation set. Five highly robust and scalable machine learning binary classifiers were used to develop the predictive models that were trained with features like molecular descriptors and fingerprints and then validated rigorously to achieve acceptable performance in terms of a set of performance metrics. The mean AUC scores for all the five classifiers with the hold-out test set were obtained in the range of 0.66–0.71. The models were further used to predict the probability score for the clinical candidate dataset. The top compounds predicted to be toxic were analyzed to estimate different dimensions of toxicity. Apparently, through this study, we propose that with the appropriate use of feature extraction and machine learning methods, one can estimate the likelihood of success or failure of investigational drugs candidates thereby opening an avenue for future trends in computational toxicological studies. The models developed in the study can be accessed at https://github.com/gnsastry/predicting_clinical_trials.git.Click on the image for more details.